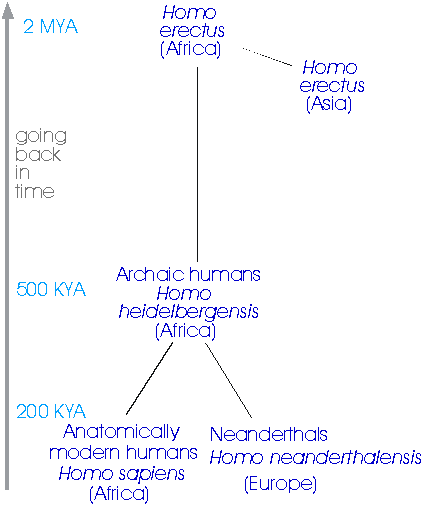
Different demographic histories, such as constant population size or expansion, create differently shaped genealogies, and we can (a) estimate the shape of the genealogy by drawing phylogenetic trees using the variation in DNA at the locus, and (b) date parts of the tree using the molecular clock. The first major example of this kind of approach was set out in a paper that came out on the 1st of January 1987 in the journal Nature, which used an analysis of the gene tree of human mitochondrial DNA to test, and support, the Out-of-Africa (OOA) theory of modern human origins. There are three kinds of human we have to deal with in this story (Fig. 6):
- Homo erectus, who evolved in Africa about 2 million years ago and dispersed rapidly throughout Asia (Peking Man and Java Man). They gave rise to:
- Archaic humans, including the Neanderthals, who were similarly widespread until they disappeared over the last 100,000 years, with the last Neanderthals surviving until about 27,000 years ago in Gibraltar.
- Anatomically modern humans, more or less indistinguishable skeletally from ourselves, first appear in the fossil record about 120,000 years ago, in East Africa, at the Kibish River in Ethiopia, and then in South Africa, at Klasies River Mouth. They appeared in Israel about 100,000 years ago, and in other parts of the world within the last 60,000 years or so.
 |
 |
| Figure 6 | Figure 7 |
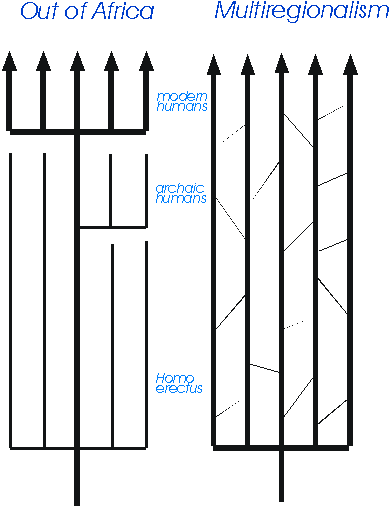
There are two main theories to explain the transitions involved (Fig. 7):
- The Multiregional hypothesis that the ancestors of modern humans, Homo erectus, spread from Africa about 2 million years ago and evolved into archaic humans (Homo sapiens) and then into anatomically modern humans (Homo sapiens sapiens) in each region - Peking Man into Chinese, Java Man into Australians, Neanderthals into Europeans and so on. Milford Wolpoff is the leading proponent of this view.
- The alternative is known as the Garden of Eden, Noah's Ark, Replacement or Out-of-Africa hypothesis. It argues that anatomically modern humans evolved from a small subgroup of archaic humans, usually thought to be in Africa about 100,000 - 200,000 years ago. This new population then expanded more recently out of Africa into other parts of the world, perhaps as a result of having acquired a more complex language, and replaced all the populations of archaic humans elsewhere, which all went extinct. In this view even the taxonomy is different: archaic humans become Homo heidelbergensis, Neanderthals are Homo neanderthalensis and modern humans are just Homo sapiens. In this country this view has been especially associated with Chris Stringer at the Natural History Museum. The debate between the two has often been very acrimonious, and both sides regularly like to accuse the other of pandering to racism, on equally feeble grounds.
mtDNA as a Molecular Marker
Cann, Stoneking and Wilson (1987) addressed the question using mitochondrial DNA (mtDNA). mtDNA was the first of the new generation of DNA marker systems. It is a small circle of DNA in the mitochondria, organelles in the cytoplasm of the cell, and is present in several copies per mitochondrion so that overall there are hundreds of copies per cell, but usually all identical. mtDNA has several very important properties that make it particularly suitable as a molecular marker:
- Maternally inherited - so the gene tree is an estimate of the maternal genealogy - tells us specifically about processes on the female side of the population history. A paternal contribution (from the sperm tail) apparently does enter the egg at fertilisation, but is somehow destroyed or lost subsequently. This also means that although there are many copies of mtDNA per cell, it is effectively haploid since they have all been inherited from the mother.
- Effectively no recombination - despite several claims, which have not been substantiated - so that there is an unbroken genealogical history. Provided there is no paternal contribution, recombination could be occurring without ever becoming visible, since the mtDNA types in one individual are all the same as each other.
- High mutation rate - about ten times nuclear DNA and even higher in the non-coding control region - so that there is a high density of mutations, allowing the main branches in the genealogy to be revealed without too much sequencing effort.
- High copy number - which raises the possibility of ancient DNA analyses, using PCR.
MtDNA is usually analysed in one of two ways - by sequencing the hypervariable non-coding control-region, or by RFLP typing the whole molecule. RFLP (restriction-fragment-length polymorphism) typing involves digesting the whole molecule (in several large chunks) with a variety of restriction enzymes, which cut at particular base sequences (such as AATT, or ATGCAT), giving a characteristic pattern called a restriction map. This latter method gives better results than just sequencing the control region, since it covers more of the DNA, but sequencing is more common as it is easier. Nowadays workers are starting to combine the approaches to give better resolved gene trees.
mtDNA Evidence for Out-of-Africa
The first OOA paper from the Wilson lab used the RFLP approach, cutting the whole molecule with 12 different restriction enzymes. They got the DNA from placentas of women in US hospitals (and Australia and New Guinea) but with ancestry all round the world - African-Americans representing Africans, East Asians, Caucasians, aboriginal Australians and New Guineans. 147 individuals in total gave 133 different RFLP haplotypes - so most were different from each other. They then ran the resulting data through a very popular maximum-parsimony program called PAUP (Phylogenetic Analysis Using Parsimony). The result has been described as "an icon in Palaeolithic archaeology" - a gene tree in which the geographical origins of the sample were indicated at the tips.
The tree became famous for one main feature: it divided into two primary branches, one including only Africans and the other containing members of all five populations. In other words, the root was within African lineages. This suggested that non-Africans had an African origin. By calibrating the molecular clock in various ways - in particular, by comparing the mtDNA diversity with the archaeologists' dates for the colonisation of New Guinea - they estimated that the common ancestor in the tree (which became known as mitochondrial Eve) was 140,000 to 290,000 years old. The out-of-Africa migration could have occurred any time between about 13,000 and 180,000 years ago. Africa seemed to be the source for human lineages not only because Africans contained the root, but also because they were the most diverse region, so it looked like they had been around for longer.
The paper led to storms of controversy. At the time, in the late 1980s, the out-of-Africa position was a minority one amongst archaeologists and physical anthropologists, and in particular these disciplines had devoted a lot of effort to establishing a place for the Neanderthals in the ancestry of modern humans. Now it looked as if the Neanderthals were an evolutionary side branch, a dead end in fact.
Criticisms of the mtDNA Work
There were immediately a number of criticisms. Firstly, there was confusion surrounding the term "mitochondrial Eve" (which had been coined by the San Francisco Chronicle). mtEve was supposed to be the woman who had harboured the most recent common ancestor of all mtDNAs in the world today, who had lived about 200,000 years ago in Africa - probably an archaic human, and probably a member of quite a large population. Her only distinction was that she was the only member of that population whose mtDNA lineage had survived down to the present day - those from everyone else alive then had subsequently gone extinct, but that didn't mean that they had not passed down genes from other loci - they may well have done. The significance was where and when she lived, not who she was. Unfortunately the name led many people, including many scientists, to think she was supposed to have been the single mother of all modern humans - that she had been the only woman alive at the time. It is not really clear whether the Wilson group themselves believed this initially. In any case, it took several years for the matter to be cleared up, and people sometimes still make the same mistake today.
Secondly there were more pointed criticisms of the methodology. Perhaps the most important were that African-Americans might not be representative of modern Africans, and that the rooting method may not have been appropriate. In a second paper, in 1991, the Wilson lab responded to these points using control-region sequence data, and using the chimpanzee as an outgroup to get a more accurate fix on the root. They got the same result. It was then that all hell really broke loose, and some of us who had just entered the field wondered whether we really ought to be doing something else.
Problems with the mtDNA Tree
This time, it was realised by several groups independently that the phylogenetic analyses in the Wilson work were insupportable. As we saw earlier, maximum parsimony programs such as PAUP attempt to find the shortest tree. Unfortunately, the tree presented by the Wilson team was not the only one possible. In fact, it seemed likely that there were at least hundreds of thousands of alternatives of the same length and the one the Wilson team published was just one taken a random. Not even at random in fact, as it turned out that it was dependent on the order the data were typed in. Even worse, it turned out that the tree they presented was not even of the shortest possible length. PAUP does both what are termed "exact" searches and "heuristic" searches. The trouble is that the number of possible trees goes up exponentially with the number of taxa. For large data sets there are just too many trees for the computer to search and one has to take short cuts and do a heuristic search instead. The problem is that a heuristic algorithm doesn't guarantee to find the shortest tree. To see the problem, imagine something called "tree space", a landscape populated by all the trees that can account for a particular data set. The tree space for about 140 mtDNA sequences is vast, but another problem is that it is structured - groups of similar trees tend to form "islands". The problem can be that the algorithm can get stuck looking on one island and not be able to find its way over to the next, so that not all the different groups of trees are explored. When others ran PAUP more carefully, they found shorter trees for the Wilson data. Again, though, there were thousands of trees of the same length, and not all of them had their roots in Africa.
These critiques came from two quarters in particular: from Maddison and coworkers, who remained neutral on whether Out of Africa was correct, and from Templeton, who believed mtDNA actually supported the Multiregional hypothesis. Out of Africa was in deep trouble for a while. In the pro camp, people fell back on the argument that Africa had the highest diversity, suggesting that the African population was older. But that didn't wash either. The diversity in a population may reflect its age if it has expanded from a very small founder population, but that was not demonstrated. Otherwise, the diversity is simply a function of the mutation rate and the long-term population size. If the human population size had been bigger in Africa for other reasons than being the source of modern humans, the effect on diversity would have been the same.
Reanalyses
However, things were not really as bad as they seemed. Most of the trees from the reanalyses still rooted in Africa. And gradually, more reanalyses of the mitochondrial data sets were performed that bore out the original conclusions. For example, in 1995, Penny and his colleagues published an extensive reanalysis which had taken 6 months of computer time to run. They repeated the parsimony analyses more than 1,000 times, making sure they explored many different "islands" of trees. They found that overall the results were similar to the Wilson group's initial claim: there was one deep cluster of Africans only, and another with both Africans and people from the rest of the world. Later on, new phylogenetic analyses with a much more comprehensive set of African sequences than before, also showed strong support for a set of deep, diverse African lineages, with just one branch leading to the whole of the rest of humanity, both Africans and non-Africans.
The only voice in the genetics community still to strongly favour the Multiregional theory was that of Alan Templeton. Templeton had done his own parsimony analyses which suggested that mitochondrial variation was not structured geographically, but was similar throughout the world. However, he was using very low resolution RFLP data where many of the most important sites that indicate regionally distributed clades had not been assayed. Furthermore, he rooted the tree on the most frequent type - which actually turned out to be the type that came out of Africa, not the most recent common ancestor. Getting the position of the root right is clearly absolutely critical, since the most important evidence for OOA relies on the root being with African lineages. The most reliable rooting method is to use an outgroup - a lineage that is more divergent than any in the group we want to root. The problem with the human mtDNA tree was that the nearest species, the chimp, diverged from us at least 5 million years ago, and the mtDNA control region evolves so fast that many of the positions would have mutated back and forth many times after such a long time, making the rooting unreliable. However, a couple of years ago ancient DNA came to the rescue of OOA.
Re-rooting with Neanderthal DNA
DNA was first extracted from extinct animals and human mummies in the mid-1980s, and looked extremely promising when PCR - able to mass-produce DNA fragments from a single copy - arrived on the scene. However, it hasn't really lived up to this early promise. DNA survival was usually very poor in prehistoric samples, and the hypersensitivity of PCR that made it all look so promising in the first place turned out to be the cause of the greatest problem - contamination by tiny amounts of modern DNA. It may be that these problems are never overcome in the case of studies of anatomically modern humans, who are not very different from their excavators and DNA-extractors, genetically.
However, in 1997, Matthias Krings and his colleagues in Munich obtained a DNA sequence from the original Neanderthal specimen from the Neander Valley, in Germany. With years of painstaking work, they had sequenced dozens of clones of tiny PCR fragments less than 100 bp long, piecing together the tiny degraded fragments of surviving DNA to reconstruct a 400-bp mitochondrial DNA control-region sequence that could be compared with that of modern humans. The result was authenticated by duplicating the sequence of one of the fragments in a second lab, but to some extent this was unnecessary: the sequence was too unusual to be simply the result of a modern contamination event. It was clearly very divergent from modern humans. Since the sequence was from the mtDNA control region, it could be used to root the mtDNA tree more accurately than before. In fact, the sequence shared nearly all of the hypothetical ancestral bases predicted for the "mitochondrial Eve" sequence type (with just one recurrent mutation at one of the sites), and as an outgroup rooted the tree more accurately than ever before. This looked like the final nail in the coffin for the Multiregional theory, at least for the maternal side of the story.
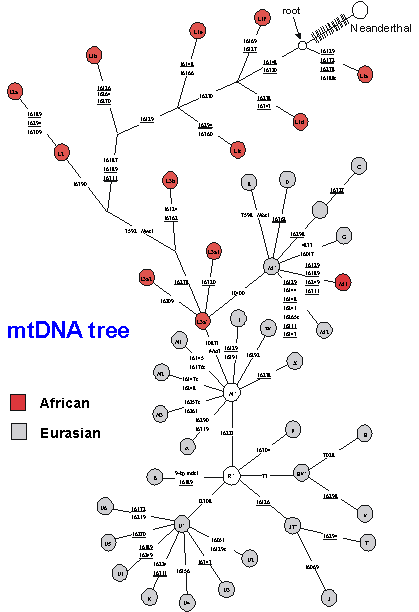
More detailed studies on the mtDNA tree, using much larger sample sizes, meanwhile, have shown that essentially the entire Eurasian variation can probably be traced back to a single African lineage, approximately 60-80,000 years ago. The overall tree for human mtDNA is shown schematically in Fig. 8: circles represent distinct haplogroups and the branches are labelled by the positions of some of the diagnostic mutation. The L3a node gives rise to the two major Eurasian haplogroups, deriving from the M and N nodes. (The M1 haplogroup appears to be a back-migrant to Africa.) Whilst diverse, deep-rooting clusters remain in Africa, with different clusters in different parts of the continent, the tree suggests that a single sequence type expanded both through eastern and western Africa and out into Eurasia. Such a drastic reduction in diversity due to the founding of a new population by migration is called a founder effect. This is again an effect of drift: the population size is dramatically reduced and the number of surviving lineages is very low. Both mtDNA and Y chromosome are especially sensitive to drift, because there are effectively four times less copies of them in the population than autosomal genes (as they are effectively haploid and only inherited through one parent). For the lineages carried into the new zone by the founding group, the molecular clock is effectively reset, allowing us to estimate the time of the founder event in the new region.
The single founding type in the mtDNA tree that spreads outside of Africa then divides into a number of clades, or haplogroups as they are known - sets of lineages with a common ancestor. There is one set of haplogroups for west Eurasians (Europeans, Near Easterners and North Africans), another set for South Asians (people from the Indian subcontinent), another set for East Asians, and another for New Guineans and Australian aborigines. People in more recently colonised areas, like Native Americans and Polynesians, have subsets of the haplogroups, suggesting more founder effects as people spread further around the world.
Other Loci
There was a final criticism to address concerning the origins of modern humans - perhaps the most demanding of all. MtDNA is only a tiny fraction of our genome. What if our mtDNAs came out of Africa, but that Homo erectus men bred with the newcomers, so that the Eurasian populations were a mixture of both? To investigate that possibility, it would be necessary to start analysing many more loci.
A good start has now been made with that. The results tend to support Out of Africa - if not yet unequivocally. We will take a look at several of the best analysed nuclear loci: the Y chromosome, an X chromosome locus, and finally ß-globin.
Y Chromosome
After mtDNA, the Y chromosome is by far the best studied genetic system in human populations. The Y chromosome is inherited down the paternal line; in this respect it complements mtDNA, although it differs in that it is passed on only to sons, whereas mtDNA is passed on both to sons and daughters but stops with the sons. So it is inherited in much the same way as surnames. Because it is non-recombining, it can be analysed genealogically in much the same way as mtDNA. Until recently it had the disadvantage that very little variation had been found - now, however, researcher have identified both slowly-evolving base variants and fast-evolving microsatellite markers on the Y. Microsatellites are repeats of short stretches of a small number of bases, such as the 2-base pair repeat CTCTCTCTCT … or the 4-base repeat ATTGATTG …
The slowly-evolving base variants, defining haplogroups as with the mtDNA, can be used to build up gene trees, and the microsatellites are useful for higher genealogical resolution and dating.
The global Y tree (Fig. 9) is still somewhat less resolved than that for mtDNA, but nevertheless has a similar distinctive structure to the mtDNA tree. As with mtDNA, the root seems to be within the African lineages. The data can, again like mtDNA, be reconciled with the emigration of a single lineage out of Africa, which then diversified as it spread round the world although this is not yet clear - but if so, this would mirror the situation in mtDNA. The root has been dated at about 190,000 years, although estimates vary widely. One curious feature is a set of lineages which seem to come out of Africa and then go back again, about 40,000 years ago, but perhaps more likely the ancestor of these types will be eventually found somewhere in East Africa. Overall then there seems to be agreement with the OOA theory from the Y.
 |
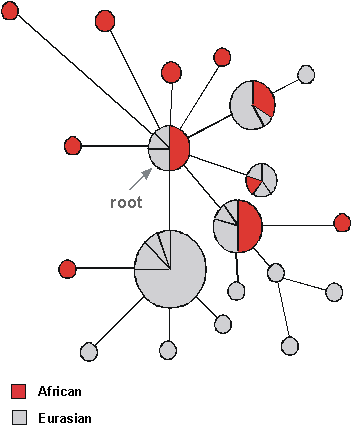 |
| Figure 9 | Figure 10 |
X Chromosome Locus Xq13.3
As we said earlier, selection for an advantageous mutation can give a similar starlike tree to that resulting from a population expansion. That means it can be very hard to distinguish selection from expansion when looking at a single locus, and, although it is encouraging that they give similar results, data from both Y and mtDNA are susceptible to the criticism that they might be the result of selection.
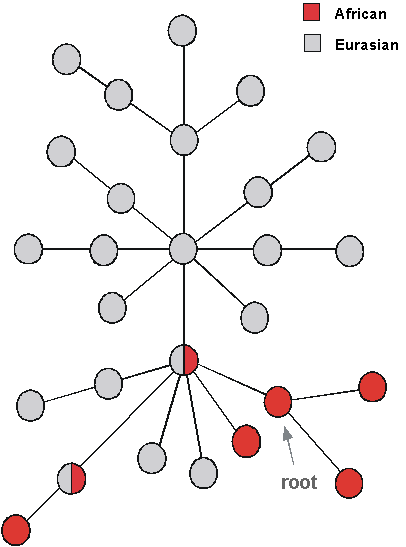
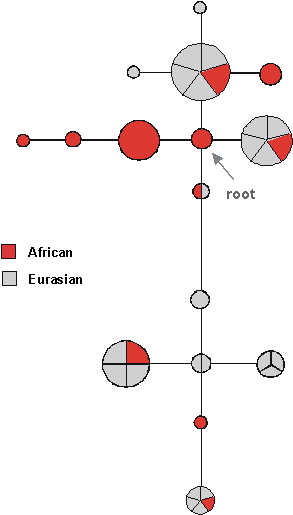
One way to get round this problem would be to look at recombining autosomal DNA sequences - but then the problem is that recombination tends to destroy genealogies by breaking up the alleles. Ideally one would like a non-coding region that more or less lacks recombination, but which is flanked by recombining sequences so that it is not susceptible to selection. An attempt to analyse such a region has been made by a team from Munich, looking at a 10kb region of the X chromosome which was both non-coding and had a very low recombination rate (Fig. 10).
Here the root, identified again using the chimp, is a type with both African-only branches and some which leading to both Africans and non-Africans. Interestingly, the root sequence - the sequence of the MRCA of the entire sample - also appears to be one of the OOA lineages. This is about 500,000 years old, whereas the OOA age would be much more recent. However, here it looks as if about four X lineages came out of Africa, compared with one each for the Y and mtDNA - a nice result, since the effective population size of X is three times that of Y and mtDNA. To think about this, consider that there are four copies of any autosomal locus compared to mtDNA - two each in both men and women - compared to one of mtDNA (in the woman) and one in the Y (in the man). For the X, there are two copies in women, but only one in males, making three overall for every four autosomal genes. As we said before, drift depends solely on the effective population size - and during the emigration from Africa, it looks as if the population size was very small indeed, so that the effect of drift was enormous. This again supports the idea of a strong founder effect during the migration out of Africa.
So far, the genetic support for OOA seems to be pretty strong. There is evidence at a number of loci - a few more could be mentioned - that a small subset of Africans set off from East Africa between 50,000 and 100,000 years ago and spread across Eurasia.
However, there are a few flies in the ointment, which we'll look at finally before moving on.
The ß-globin locus
A similar global study of variation, this time looking at 3kb of the ß-globin locus, was carried out in Oxford and published in 1997. Here there was some recombination going on, the probable recombinants were screened out before doing a genealogical analysis. This was one of the first papers to be published that performed a full statistical treatment on the data using the new tools of population-genetics theory.
The most obvious feature of the ß-globin gene tree is that it is not at all star-like, and indeed there is no signature of an expanded population (Fig. 11). Perhaps the most likely explanation for this is that, because the mutation rate is very low, we are looking back further in time, so that most of the mutations occurred before the human population had expanded: the coalescence time of the tree overall is about 750,000 years.
What is particularly startling, however, is that one of the clades, which occurs throughout Eurasia and is very rare in the African samples, is estimated to be more than 200,000 years old. The authors interpreted this as substantiating the idea that there was some breeding with archaic human populations after all - one of the descendants of Homo erectus still living in East Asia 200,000 years ago.
Still, this isn't the only possible interpretation. The culprit clade is widespread throughout Eurasia, in contrast with the more restricted patterns of mtDNA and the Y chromosome which indicate quite limited gene flow between east and west. So it seems a bit unlikely that this could be signalling limited mixing between newcomers from Africa and archaic humans in East Asia 50,000 years ago, say, which would not be detected at other loci. Moreover, the clade is not entirely absent from Africa. It is perhaps possible that it has been reduced by drift in the African populations examined, or simply been missed in the sampling process - Africa is a big place, and the number of samples tested very limited. It would be very exciting to find genetic evidence for inter-breeding between modern and archaic humans during the expansion out of Africa, but more evidence will be needed for most people to be convinced.
So overall, the evidence does point to a strong founder effect as a small group of Africans expanded into Eurasia 50,000 to 100,000 years ago. This seems reasonably consistent with the archaeological record, which signals a major, sophisticated new set of stone tools, art, and social relations, starting about 50,000 years ago - possibly earlier in Africa.
The Settlement of the Pacific
We have seen how the genealogical approach has helped to clarify our views of modern human origins. Nowadays, this approach is being employed to investigate a whole range of questions about the subsequent spread of modern humans into different parts of the world. To finish off, we'll take a look at an example of these: the settlement of the Remote Pacific Ocean, within the last 4,000 years.
Modern humans - certainly using boats - settled the western part of the Pacific, close to New Guinea and Australia, more than 30,000 years ago. However, in the last 4,000 years, people spread further, settling the thousands of small islands right across the Pacific that we call Polynesia. This is an attractive model system for trying out the genetic approach, because we have quite a lot of evidence from archaeology and linguistics with which to check the genetic data. For example, there is radiocarbon evidence for the first settlement of many islands in Polynesia that suggests an expansion from the western Pacific starting about 3,500 years ago, and ending up with the colonisation of New Zealand about 1,000 years ago. Although the archaeology is more equivocal about where the people ultimately came from, they all speak Austronesian languages, which occur today throughout Island Southeast Asia and many parts of Melanesia.
We can split the competing hypotheses on Polynesian origins into two main groups: they cam either from (1) Southeast Asia or (2) New Guinea/Melanesia. There has also been the suggestion of Thor Heyerdahl that they came from South America, which has never been taken very seriously since both archaeology and linguistics suggest and origin from the west.
The mtDNA picture for the Polynesians is extremely simple (Fig. 12). They have two main clades (or haplogroups), which are extremely divergent from each other, suggesting very different origins. One clade accounts for about 94%, and the other for nearly 4%. Although they are very different from each other, neither clade is very diverse internally. An estimate of the age within Polynesia of the commonest clade comes to about 1,000 years within central Polynesia and 3,000 years in the west, matching the archaeological dates very well.
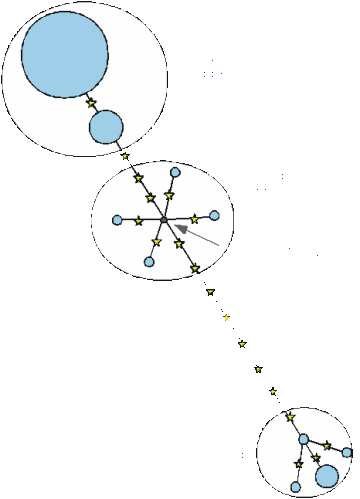 |
 |
| Figure 12 | Figure 13 |
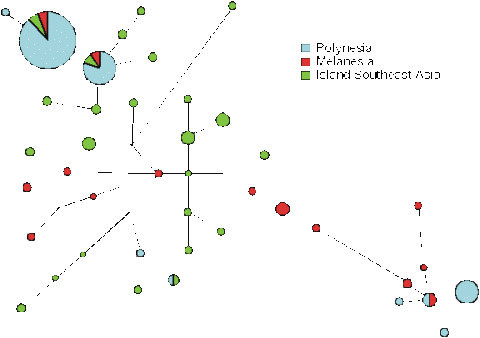
Now, we can look to see how these clades fit into the overall tree of variation for Southeast Asia and the western Pacific (Fig. 13). Not surprisingly, the overall tree is far more diverse, and Southeast Asians and Melanesians are quite distinct, falling onto different branches. We can see that the main Polynesian clade emerges from the tip of one of the Southeast Asian branches, and the minor Polynesian clade comes from the opposite end of the tree, on a branch present only in Melanesians and New Guineans. So it looks as if the majority of Polynesian mitochondria cam from Southeast Asia, with a small minority coming from the western Pacific - perhaps being picked up on the way. The remaining 2% of lineage in Polynesia are of either Asian or Europe origin, as one might expect - there seem to be none from Native Americans.
We can even go further, because there is a difference between archaeologists who, whilst agreeing that the Polynesians arose in Southeast Asia, differ on whether it was from Taiwan and the South Chinese mainland, or somewhere within the islands of Indonesia. The main Polynesian sequence doesn't occur in Taiwan, but it does occur in Indonesia. Since it occurs nowhere else to the west, it looks as if it must have arisen there, and then spread west. It has a lot of diversity in Indonesia, amounting to something like 10,000 to 20,000 years, so it looks as if the idea that the Polynesians arose in Indonesia is likely to be correct.
In summary, we can learn quite a lot about prehistoric migrations by looking at trees built up from DNA sequences, and seeing how the lineages on the trees are distributed geographically in present-day populations. Usually we need to start off with an overall picture of the demography - whether the tree is star-like, signalling an expansion - because of there have not been expansions and drift has played a large role then tracing lineages back and especially dating clades becomes more problematic. Fortunately most human populations do show signatures of demographic expansions, so we can proceed to study the movements of lineages in more detail. The situation is not usually as straightforward as the expansion into Polynesia, and the subject is really still in its infancy. Moreover, most of this work has been done up till now using mtDNA, showing only the female side. However, the Y chromosome is starting to give us a complementary picture of the male side, and eventually more information from non-recombining parts of the X and autosomal loci will help to build up a fuller picture.